Executive Summary: The Document Bottleneck Between Trial Completion and Regulatory Submission
Your database lock date just passed. An AI medical writing automation solution addresses exactly what comes next. The process of extracting values from SAS outputs, populating regulatory templates, and reconciling tables against narrative text consumes weeks of highly qualified writer time. All of this unfolds before a regulatory submission window even opens.
Rather than requiring writers to transcribe clinical data by hand, the system reads structured statistical outputs directly and generates source-tagged regulatory narrative. The underlying architecture separates fact retrieval from text generation – ensuring every number traces to its validated source record before a writer reviews a single sentence. For pharmaceutical companies, CROs, and medical affairs teams under continuous submission pressure, this is an operational shift that leading development organisations are already deploying to reclaim weeks from document production cycles.
1. Why Do Regulatory Document Timelines Keep Slipping Despite Larger Writing Teams?
Regulatory document bottlenecks persist because volume, complexity, and compliance demands grow faster than any team can manually absorb.
The Regulatory Writing Environment in Pharmaceutical Development
Regulatory submissions rest entirely on the quality and timeliness of clinical documentation. A Clinical Study Report (CSR)A comprehensive document describing the design, methods, results, and clinical significance of a completed clinical trial, required for regulatory approval submissions worldwide can run 500 to 1,500 pages, drawing on hundreds of statistical tables, patient data listings, and protocol references that must cross-check consistently throughout every section. Medical writers who produce these documents combine deep scientific literacy with regulatory expertise – a profile that takes years to develop and commands significant compensation in a market where qualified specialists are scarce.
Global demand for qualified writers continues to outpace academic training pipelines, creating a structural shortage that limits throughput regardless of how many teams a sponsor organisation assembles. The problem compounds on large development programmes where multiple studies complete simultaneously, creating parallel CSR demand that overwhelms fixed writing capacity.
In practice, organisations deploying AI to regulatory writing teams consistently encounter a counterintuitive finding: the bottleneck is not the prose drafting itself, but the data extraction and cross-referencing that precedes it. Automating that upstream layer – before a writer touches the document – delivers larger time savings than automating narrative generation alone.
Compared to fully manual workflows, an AI-assisted approach eliminates the transcription layer entirely. Manual processes require a writer to locate a value in a SAS listing, type it into a narrative sentence, and then verify it again during review. An automated regulatory writing software system retrieves the value, embeds it in a generated sentence, and tags it with its source record – reducing that task from minutes to seconds while removing the human transcription error risk completely.
Key Pain Points This AI Solution Addresses
- Medical writers spending too long on routine drafting – highly skilled professionals dedicate the majority of their time to mechanical data extraction rather than the scientific interpretation and clinical reasoning that genuinely requires their expertise
- Regulatory document deadlines being missed – multiple concurrent submissions competing for limited writer capacity routinely push timelines past planned regulatory windows, with direct commercial consequences for pipeline assets
- Inconsistent document language across different writers – terminology, phrasing conventions, and formatting standards vary person to person, creating reviewer burden and downstream cross-document consistency problems
- High cost of outsourcing medical writing – engaging CRO writing teams or freelance specialists for overflow volume represents significant and growing expenditure that scales poorly with portfolio growth
- No automated way to generate patient narratives from adverse event data – safety reporting requires individual narratives for hundreds or thousands of patients, all constructed manually from structured safety datasets
- Clinical writing team acting as a bottleneck – regulatory submissions cannot advance until documents clear the writing queue, making the writing function a rate-limiting factor in the entire development and approval timeline
- Document quality inconsistent across different submissions – without systematic terminology and style controls, quality varies between programmes, writers, and time periods in ways that create rework at review stage
Why Traditional Approaches Fall Short
- Manual extraction from SAS datasets introduces transcription errors that cascade across the document and require expensive rework cycles to resolve at review stage
- Copy-paste workflows between statistical output and narrative text create version control risks whenever statistical data undergoes late revisions – a common occurrence in Phase III programmes
- Scaling output by hiring additional writers does not solve the structural speed problem: it multiplies cost without addressing the underlying data-to-text conversion bottleneck
- Freelance overflow writing introduces variable style and terminology that creates an editing burden for in-house writers who must standardise language before submission
- Human review for consistency across 800-page documents takes almost as long as initial drafting, because no systematic mechanism exists to flag internal discrepancies automatically
- Applying general-purpose AI medical writing software without a retrieval-locked architecture carries unacceptable hallucination risk in a regulatory context where a single factual error can delay approval
2. What Does an AI Medical Writing Automation Solution Actually Do?
An AI medical writing automation solution converts structured clinical trial data into traceable, human-reviewable regulatory document drafts – without requiring writers to manually extract or transcribe a single value.
How the System Changes the Writer’s Role
The system reads directly from validated clinical trial databases and statistical output files. It assembles the relevant facts for each document section according to the applicable regulatory template and generates narrative text with every value linked to its source record. The result reaches the medical writer as a reviewable draft – not a finished document. The writer’s role shifts from data retrieval and transcription to scientific oversight and editorial judgment – exactly the work that requires their expertise.
This AI pharma document automation platform approach does not replace medical writers. It removes the mechanical layer of their work so their capacity concentrates where it creates the highest value.
The Core Architectural Principle: Constraint Over Generation
The critical architectural principle distinguishing a reliable medical writing automation platform from a general-purpose AI tool is constraint. The language model receives only pre-verified, source-tagged facts retrieved from the study’s own validated datasets. It arranges and articulates those facts into regulatory prose – it does not generate or infer values independently. That single design constraint is what makes the output trustworthy enough for a regulatory submission environment.
Vision and Objectives
- Reduce first-draft production time for standard regulatory documents from weeks to days, without compromising accuracy or regulatory compliance
- Eliminate manual data transcription as a source of error by replacing copy-paste workflows with direct database-to-text generation with full source linkage
- Ensure complete traceability – every AI-generated sentence links to its specific source data record, satisfying regulatory inspection requirements at the sentence level
- Maintain writer control over scientific interpretation, risk-benefit assessment, and final submission sign-off throughout the entire document lifecycle
- Scale document output capacity to match portfolio growth without proportional headcount or outsourcing cost increases
- Deliver consistent terminology, regulatory style, and structural compliance across all documents within a programme
3. How Do Real Pharmaceutical Teams Apply This AI Solution in Practice?
The following scenarios show this clinical writing AI solution in action across different document types, team sizes, and operational pressures.
Large Pharma Programme: CSR First-Draft Acceleration
Your trial hit database lock six weeks ago. The TLFs package has been final for four weeks. But your CSR first draft is still six weeks away – and the submission window is fixed. A Phase III programme running multiple concurrent studies faces this situation repeatedly across its development timeline. Each CSR demands the same intensive data extraction, template population, and internal consistency review, with no mechanism to parallelize the work beyond writer headcount. The AI medical writing automation platform connects directly to the statistical output package, interprets the tables and listings, and auto-populates the ICH E3 document structure with source-linked narrative text. Efficacy summaries, patient disposition, and safety overview sections appear as tracked changes in the writer’s authoring environment. The writer reviews and approves scientific framing rather than constructing sentences from raw data. The first reviewable draft reaches the team in days rather than weeks.
CRO Safety Team: High-Volume Patient Narrative Generation
Your safety team needs 400 individual patient narratives submitted before the safety data cutoff – and two medical writers are assigned to the trial. At two to four hours per narrative using manual methods, that workload is incompatible with any realistic timeline this team can absorb. The automated patient narrative generation tool reads each adverse event record directly from the EDC system, applies the sponsor-approved narrative template, and generates a source-linked draft for each patient record. Writers review generated narratives rather than drafting them individually. The same 400 narratives that would have required weeks of concentrated effort now complete in a fraction of the time – freeing the safety team to focus on causality assessment and the medically complex cases that genuinely require clinical judgment.
Medical Affairs Team: Annual DSUR Preparation
Your annual DSUR (Drug Safety Update Report)An annual regulatory document summarising the safety profile of a medicinal product in clinical development, submitted to regulatory authorities under ICH E2F guidelines is due in 45 days, and your medical affairs writer needs the full period just to compile the aggregate safety data review. This is a recurring pressure point across active development programmes where safety data accumulates from multiple sources throughout the year. The AI regulatory writing solution aggregates data from safety line listings, literature search results, and cumulative safety data files, then populates DSUR template sections with generated narrative text linked to each contributing source. The first draft of the annual safety summary is ready within days of data availability – giving the medical affairs team weeks of review and scientific editing time rather than weeks of document construction.
Ready to explore what this solution looks like for your organisation?
Talk to Our AI Team4. How Does an AI Medical Writing Automation Solution Process Clinical Data Into Regulatory Drafts?
The system moves clinical data through a structured pipeline that separates retrieval from language generation – the architectural decision that makes accurate, traceable regulatory output possible.
Data Acquisition: What the System Consumes
The system connects to clinical trial data repositories across the full data landscape. Source inputs include EDC (Electronic Data Capture)Software systems used to collect and manage clinical trial data electronically, replacing paper-based case report forms and serving as the primary source of patient-level trial data systems, SDTM (Study Data Tabulation Model)The CDISC-standard format for organising and formatting clinical trial data for regulatory submission to FDA, EMA, and other agencies datasets, ADaM (Analysis Data Model)The CDISC standard for analysis-ready datasets derived from SDTM, used to produce the statistical outputs that form the numerical basis of clinical study reports files, and complete statistical output packages – the TLFs (Tables, Listings, and Figures)The statistical tables, patient data listings, and graphical figures generated from clinical trial analysis, which form the numerical backbone of every CSR and regulatory dossier package that underpins every regulatory document.
Source documents including the clinical protocol, statistical analysis plan, and applicable regulatory templates are also ingested. These provide the structural and contextual framework that governs how generated narrative maps to each document section.
The AI Processing Pipeline
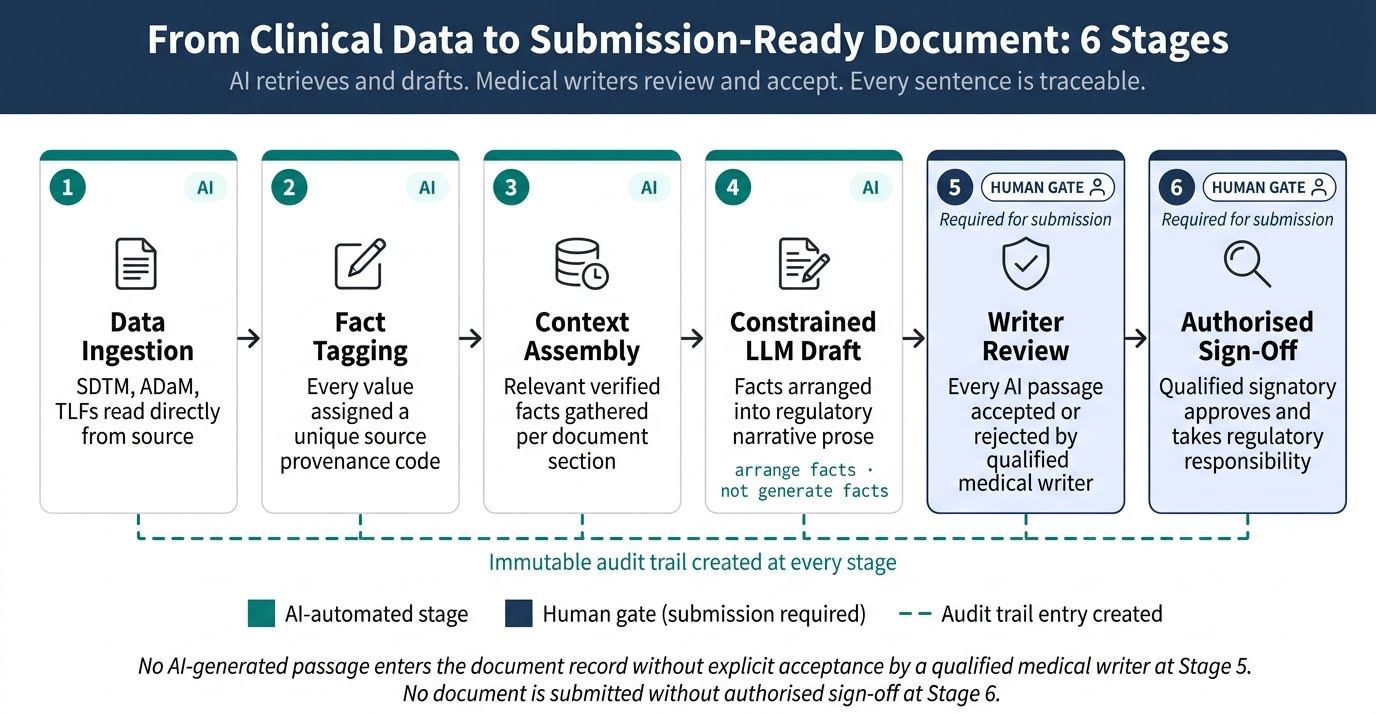
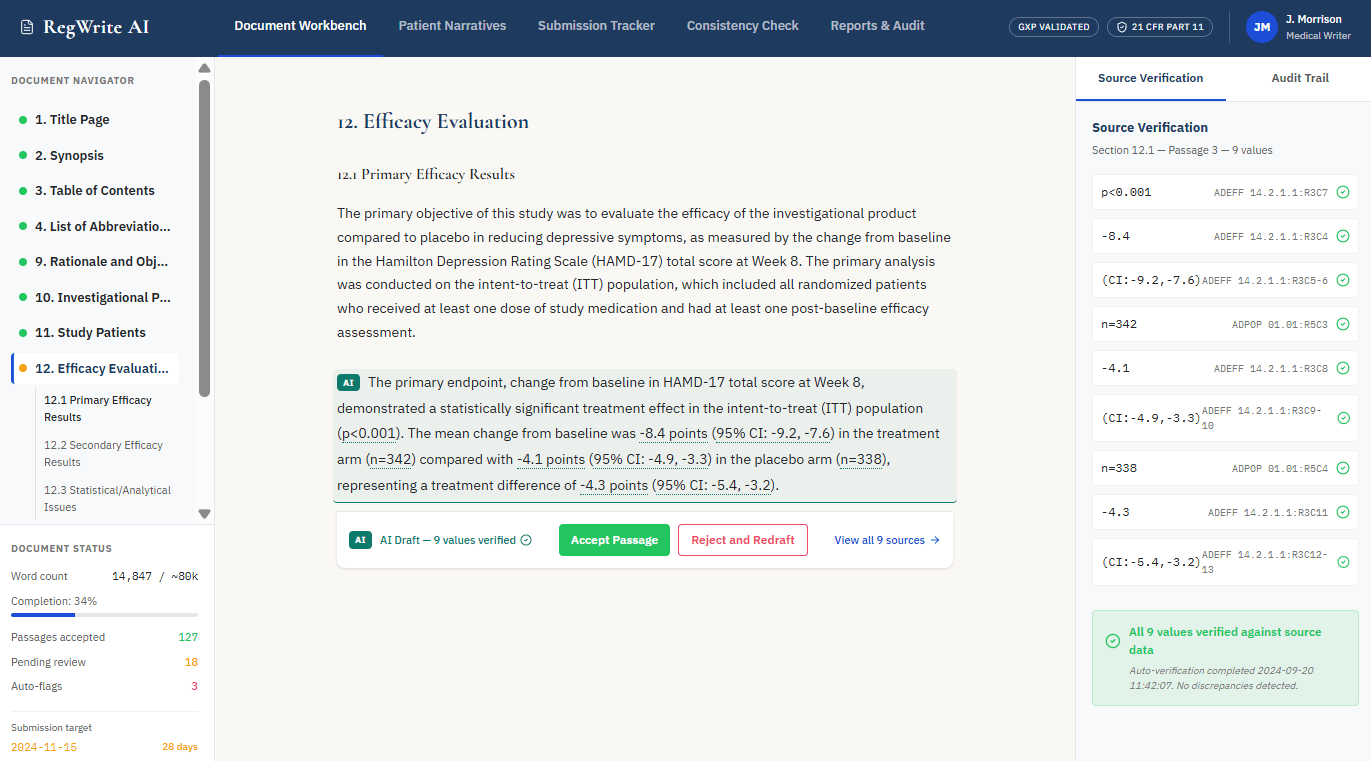
- Data Ingestion and Structured Extraction: First, the system connects to the trial’s data repositories and extracts numerical values, adverse event records, patient demographics, and efficacy endpoints from validated source files. Every extracted value carries metadata identifying its precise source table, column, row, and file version – establishing the data provenance layer before any text generation begins. Nothing enters the pipeline without a confirmed source address.
- Fact Tagging and Source Indexing: Next, each extracted value receives a unique source tag that maps it to its exact origin in the validated dataset. This indexing step is the architectural core of a reliable AI regulatory document drafting tool. The language model that follows receives only pre-tagged, source-verified facts – not open access to its training data or the ability to infer values it did not retrieve from the study’s own data sources.
- Deterministic Retrieval and Context Assembly: The retrieval engine then pulls the relevant, pre-tagged facts for each document section according to the regulatory template structure. For a CSR section covering primary efficacy results, for example, the engine assembles the specific endpoint values, patient disposition numbers, and statistical test results that belong in that section. This bounded, verified context – not a general LLM prompt – becomes the sole input for text generation.
- Constrained Language Model Drafting: A fine-tuned LLM (Large Language Model)An AI language model that has been further trained on domain-specific regulatory and clinical text after initial general training, improving its accuracy and style conventions for pharmaceutical regulatory content converts the assembled, source-tagged facts into compliant regulatory narrative. The model operates under strict output constraints: it arranges and articulates pre-verified facts into prose, but it cannot generate or infer numerical values independently. A common pattern across real implementations of this solution is that this single constraint – “arrange facts, do not generate facts” – is what separates production-ready regulatory AI from general-purpose tools that hallucinate in clinical contexts.
- Automated Cross-Reference Verification: Once the draft is generated, a verification agent re-reads every numerical value in the output and checks it against the source index. Any discrepancy between a generated number and its tagged source triggers an automatic flag before the document reaches a human reviewer. This layer provides systematic cross-reference checking that manual review workflows cannot replicate at the same speed or consistency.
- Human Review Interface and Audit Trail: The verified draft surfaces to the medical writer as tracked changes within their existing authoring environment – not as accepted final text. The writer reviews, accepts, or rejects each AI-generated passage explicitly. Every decision logs with a timestamp and user identity, creating a sentence-level audit trail that supports regulatory inspection readiness from the moment drafting begins.
Human-in-the-Loop: Where Human Judgment Still Matters
- Scientific interpretation of results: the AI generates factual narrative from structured data; the medical writer determines what the results mean clinically and how to frame them for a regulatory audience
- Risk-benefit assessment and discussion sections: these require qualified medical judgment that extends beyond structured data and cannot be delegated to automated generation under current technology or regulatory expectations
- Adverse event causality assessment: determinations about the relationship between a study drug and an adverse event remain the exclusive responsibility of qualified clinical experts
- Final document sign-off and submission authorisation: no automated system submits independently; a qualified signatory reviews, approves, and takes full regulatory responsibility for every document
- Edge cases and flagged passages: the system escalates sections where source data is ambiguous, incomplete, or conflicts with protocol expectations – rather than attempting to draft through uncertainty autonomously
Output and Interaction: How Results Reach the Writer
The system delivers output through interfaces writers already use rather than requiring migration to a new platform. Tracked-change Word documents appear in the writer’s existing authoring environment, with every AI-generated passage requiring explicit acceptance before it becomes part of the document record. Direct API connectors to regulatory content management systems allow generated drafts to appear within existing version control and approval workflows. Automated consistency reports flag terminology variations across sections. Progress dashboards show drafting completion status against submission timelines – giving project managers visibility without changing the writer’s working environment.
5. What Technologies Power an AI Clinical Document Writing Tool?
Each technology in this stack serves a specific function – understanding what each one does helps evaluate whether a given implementation will perform reliably in a regulated pharmaceutical environment.

- Large Language Models (LLMs) fine-tuned on regulatory corporaAI language models further trained on ICH guidelines, clinical protocols, and approved submission language to produce output that matches pharmaceutical regulatory style and structural conventions: General-purpose language models perform poorly on regulatory text without domain-specific training. Fine-tuned models trained on ICH guidelines, clinical protocols, and historical submission language produce output that matches the style and terminology conventions regulators expect.
- Natural Language Generation (NLG)AI techniques that convert structured data – numbers, tables, and categorical values – directly into human-readable narrative text, providing highly reliable output for data-heavy regulatory document sections: NLG handles the deterministic conversion of structured numerical data into compliant narrative sentences. For patient narratives and summary table sections where output derives almost entirely from structured source data, NLG provides the most reliable and consistent generation approach in the technology stack.
- Retrieval Augmented Generation (RAG)An AI architecture that retrieves specific verified information from a defined, bounded data source before generating text – preventing the model from using its training data to invent facts outside the retrieved context architecture: RAG is the critical mechanism controlling hallucination risk. Rather than allowing the language model to draw freely on its training data, the retrieval layer supplies only verified, source-tagged facts from the study’s own validated datasets – bounding what the model can include in generated output.
- SDTM and ADaM data connectors: Pre-built connectors to CDISC-standard data formats allow the system to read trial data without manual export or reformatting steps. This removes a significant source of delay and error from the data ingestion phase and enables direct connection to the statistical analysis environment.
- Sentence-level provenance tagging: Every AI-generated sentence carries embedded metadata linking it to the specific source table cell, file version, and timestamp that produced its numerical content. This level of traceability exceeds what most manual regulatory writing workflows can provide.
- Private and on-premise LLM deploymentRunning the language model on infrastructure controlled entirely by the organisation – either on-site servers or a dedicated private cloud environment – so clinical trial data never passes through shared third-party APIs: Clinical trial data contains patient-level information protected under applicable privacy regulations. Deployment on private infrastructure or within an enterprise cloud configured with appropriate data processing agreements ensures this data never passes through a shared external API endpoint.
- Document management system API connectors: Integration with regulatory content management platforms via published APIs enables the system to operate within existing document lifecycle workflows. Writers and reviewers interact with generated content through familiar tools rather than a separate platform interface.
- GxP-compliantConforming to Good Practice regulations (GCP, GMP, GLP) governing pharmaceutical development processes, requiring validated systems, complete audit trails, and documented change control for all regulated activities audit trail infrastructure: Regulated pharmaceutical environments require complete documentation of every system action, user decision, and document change. 21 CFR Part 11 establishes the FDA’s requirements for electronic records and electronic signatures – the framework within which any regulated AI system handling submission documents must operate. Audit trail systems built to these validation standards make the AI system inspection-ready from the point of production deployment.
6. What Results Does AI Medical Writing Automation Actually Deliver?
AI medical writing automation delivers measurable improvements across time, quality, and document production capacity – with each benefit tied directly to a documented pain point in the regulatory writing process.
What an AI clinical document writing tool uniquely provides is not just faster output but structurally different output. Narrative is generated from verified source data rather than from manual transcription. Traceability is built in from the first sentence rather than added during review.

- Significant reduction in first-draft production time: Direct database-to-text generation eliminates the data extraction and transcription phase that typically accounts for the largest share of initial drafting time. Merck’s validated deployment reduced first-draft time from 180 hours to 80 hours – a 56% reduction – and compressed the calendar timeline from two to three weeks down to three to four days.
- Elimination of manual transcription errors: When the system retrieves values directly from source data rather than relying on human copy-paste, the entire class of transcription errors – transposed digits, misread decimal points, wrong table references – disappears. The same Merck deployment reported a 50% reduction in errors spanning data accuracy, messaging, citations, terminology, and typography. Reviewer query volumes drop as a direct result.
- Scalable patient narrative output at volume: The automated patient narrative generation capability turns what was a multi-week bottleneck for large safety datasets into a process measurable in hours. Safety teams that previously could not absorb high-volume narrative workloads can now process them without compromising submission timelines.
- Consistent terminology and style across the programme: Because all documents draw on the same templates, terminology controls, and generation logic, language consistency across a multi-study programme improves systematically – reducing reviewer rework and cross-document inconsistency findings at every review cycle.
- Writer capacity redeployment to high-value work: Medical writers freed from mechanical extraction and transcription apply their expertise to scientific interpretation, benefit-risk analysis, and regulatory strategy – the work that delivers the highest value and that only they can do. Teams increase high-value throughput without increasing headcount.
- Reduced dependency on external writing resource: Higher internal throughput reduces the volume of work directed to CRO writing teams and freelance specialists, with a corresponding reduction in associated cost and coordination overhead over each programme cycle.
- Improved regulatory inspection readiness: Sentence-level audit trails and source provenance tagging produce documentation of the writing process that exceeds most manual workflow standards – supporting regulatory authority inspection with complete, structured traceability.
- Earlier submission and accelerated commercial timeline: Faster document production compresses the interval between database lock and regulatory submission. For a development asset with significant peak sales potential, each week recovered from the writing cycle translates directly to earlier market access and measurable revenue acceleration.
7. Is an AI Medical Writing Automation Solution Worth the Investment?
An AI medical writing automation solution generates measurable return through time compression, error reduction, and the commercial value of earlier drug approval – and the business case builds differently depending on document volume and asset value.
Teams that have worked through full deployment consistently find that the ROI calculation anchors on five key business metrics, each measurable before and after implementation. Presenting these metrics with before-and-after benchmarks is how regulatory operations leaders build the internal investment case most effectively.
Key Metrics for the Internal Business Case
- First-draft production time per document: Measure hours from data availability to first reviewable draft, pre- and post-implementation. This is the most directly attributable efficiency gain and the simplest metric to track and report to leadership.
- Reviewer query volume per CSR: Track the number of data-related queries raised during document review cycles. A reduction in this number indicates a direct reduction in review burden and rework cost – both attributable to improved data accuracy in AI-generated drafts versus manually transcribed ones.
- Writer output capacity per programme cycle: Count documents completed per writer per quarter before and after AI assistance. Capacity gains translate to either cost avoidance on external writing spend or portfolio expansion without headcount addition – both of which appear clearly on a cost-benefit analysis.
- Cost per completed document: Aggregate writer time, review cycles, and external resource spend per regulatory document. This metric makes the investment case visible to finance and senior leadership in terms they recognise.
- Submission timeline adherence rate: Measure the proportion of planned submission dates met on schedule. Improvement connects the writing function directly to pipeline value – particularly material for programmes approaching patent expiry windows where every week matters commercially.
Realistic Implementation and Payback Timeline
For a mid-size pharmaceutical company or CRO, a realistic implementation timeline runs three to six months from engagement to production-ready deployment for the first document type. Patient narratives are the recommended starting point – they represent the highest-volume, most structurally consistent document type, and therefore produce the fastest measurable return on investment. Broader CSR section automation follows as the system is validated and integrated into existing workflows.
Payback timelines depend substantially on document volume. Organisations completing fewer than five major regulatory documents annually will see more modest returns relative to implementation cost. Teams managing ten or more submissions per year, or those running large safety datasets requiring hundreds of individual patient narratives, typically recover implementation costs within the first full operational year.
The most compelling rationale for acting now rather than waiting relates to compounding competitive dynamics. Every submission cycle completed with fully manual writing methods represents a fixed opportunity cost. McKinsey estimates that accelerating CSR timelines by approximately 40 percent increases NPV per asset by roughly $15 million to $30 million. That figure compounds across a multi-asset pipeline. Regulatory teams that have already integrated an AI pharma document automation platform are compressing document timelines their competitors are not – and that advantage widens with each programme cycle that passes.
8. What Does Implementing an AI Medical Writing Automation Solution Actually Require?
Successful deployment requires addressing six practical dimensions before a production system goes live – each is manageable with appropriate expertise, but none should be underestimated during project planning.
- Clean, structured source data as a prerequisite: The system performs reliably when source datasets conform to CDISC standards and maintain consistent structure throughout. Messy, non-standard, or incomplete SDTM datasets degrade output quality significantly. Data readiness assessment – reviewing SDTM conformance, variable coding consistency, and completeness – should precede technical implementation planning.
- Private infrastructure or compliant cloud deployment: Clinical trial data containing patient information cannot legally pass through public LLM APIs under most regulatory jurisdictions. Deployment via private LLM infrastructure or within an enterprise cloud environment with appropriate data processing agreements is a technical and legal prerequisite – not a configuration option.
- Regulatory system validation: Automated regulatory writing software used in the preparation of submissions operates as a regulated software system and requires formal validation under applicable GxP requirements. This includes installation qualification, operational qualification, and documented change control procedures. Budget three to four months for validation activities beyond the technical development timeline.
- Document management system integration: The system needs to operate within existing document lifecycle infrastructure – version control, approval workflows, and audit trail requirements. Integration complexity depends on the incumbent platform and the availability of published APIs. Organisations on standard platforms with open API access encounter significantly lower integration overhead than those on legacy or heavily customised systems.
- Medical writer training and change management: Technical deployment without writer adoption is not a functioning system. Writers need training on the review interface, on how to evaluate AI-generated passages against source data, and on how their accept or reject decisions build the audit record. Organisations that involve medical writers in design and piloting see substantially faster adoption than those that train after deployment is complete.
- Ongoing model and template maintenance: Regulatory guidance evolves. Template structures change as ICH guidelines are revised and agency expectations develop. The generation logic and document templates within the system require corresponding updates over time. Treat the solution as requiring periodic maintenance cycles rather than a one-time deployment event.
Where This Solution Has Real Limits
What implementation experience reveals that theoretical explanations often miss is that no current AI medical writing system handles every regulatory document type with equal reliability. The following limitations apply to the category as it stands today and should be factored honestly into any deployment plan.
- Reasoning and discussion sections require human drafting: AI generates factual narrative from structured data reliably. However, integrated risk-benefit analysis, cross-study comparison discussion, and sections requiring synthesis of evidence across a development programme still require qualified human authors who can weigh clinical context, regulatory precedent, and scientific judgment.
- Low-volume operations see limited return: For organisations completing fewer than five major regulatory documents per year, the implementation and validation overhead may exceed the efficiency gain within a reasonable payback period. This solution scales effectively with volume – its value proposition is strongest where document throughput is high.
- Data quality problems propagate faithfully: The system generates narrative from whatever data it retrieves. When source data contains errors – mislabelled records, protocol deviations not clearly flagged, or non-standard variable coding – the generated narrative reflects those errors. Data quality problems do not disappear; they surface more visibly in the output.
- Integration complexity is routinely underestimated: Legacy document management environments, custom regulatory content management configurations, and non-standard SDTM implementations all add integration time and cost that standard project timelines rarely account for fully. Early technical discovery of the integration landscape is essential.
9. Who Benefits Most from an AI Medical Writing Automation Solution?
This solution delivers highest value where document volume is high, writer capacity is constrained, and submission timelines carry direct commercial consequences.
Primary Buyer Profiles
Primary beneficiary profiles include large and mid-size pharmaceutical companies managing multi-study development programmes, contract research organisations running concurrent trials for multiple sponsors, and medical affairs teams responsible for annual safety reporting across active product portfolios.
Regulatory affairs professionals managing submission timelines and writer resource allocation represent the primary decision-making audience for this type of clinical writing AI solution. Medical writers and clinical scientists are the direct users whose daily workflows the system transforms most immediately.
This Solution Is Particularly Valuable If…
Organisations seeking to reduce their reliance on expensive external resource and build internal AI medical writing software capability will find this solution particularly applicable, especially where structured CDISC-compliant data infrastructure already exists.
- Your organisation completes more than five major regulatory documents per year and experiences writer capacity constraints at peak submission periods
- Your safety teams generate high volumes of patient narratives for clinical trial submissions and find the current manual workload incompatible with submission deadlines
- You rely on significant external writing expenditure to manage overflow volume and want to build scalable internal capacity instead
- Your regulatory affairs team operates in a CDISC-compliant data environment with structured SDTM and ADaM datasets that are ready for automated ingestion
- Your clinical development pipeline includes assets where accelerated time to submission has measurable commercial value – particularly true for programmes near patent expiry or in competitive therapeutic areas
10. Frequently Asked Questions About AI Medical Writing Automation
The following questions address the most common concerns buyers raise when evaluating this solution category.
How does an AI medical writing automation system for pharma companies handle the hallucination risk?
The hallucination risk is the central design challenge in this category, and the solution lies in architecture rather than in the language model itself. A properly built system uses a retrieval-locked approach: the language model receives only pre-tagged, source-verified facts extracted from the study’s own validated datasets and cannot generate values outside that bounded context. This is fundamentally different from asking a general-purpose LLM to write about clinical trial data from its training memory. When the architecture separates fact retrieval from text generation – and an automated verification agent checks every numerical value in the output before a human sees the draft – hallucination risk on structured data sections drops to near-zero. The remaining risk area is reasoning and discussion text, where the model must synthesise rather than report. This is precisely why those sections remain under human authorship in all responsible implementations of this technology.
Can an automated clinical study report generation platform produce submission-ready documents without human review?
No – and this is not a limitation of specific platforms but a regulatory and scientific reality that applies to the category. The FDA, EMA, and other major regulatory authorities have confirmed that sponsors bear full responsibility for the accuracy of submitted documents regardless of how content was generated. Human sign-off by a qualified medical writer and a responsible regulatory signatory is mandatory throughout the review and submission process. Responsible implementations treat AI-generated content as a first draft that requires expert review – not as a finished submission artefact. The genuine value proposition is not removing human review: it is replacing the mechanical drafting phase with AI output so that human review can concentrate on scientific judgment and clinical interpretation rather than data transcription verification.
What does an AI regulatory document writing tool for submission teams actually need to work properly?
Three prerequisites matter most in practice. First, clean CDISC-standard source data – SDTM and ADaM datasets that are complete, consistently coded, and free from structural anomalies that would mislead the retrieval layer. Second, a compliant deployment environment – clinical trial data cannot legally pass through public LLM APIs under most privacy regulations, so private or enterprise cloud deployment with appropriate data processing agreements is a technical requirement, not an option. Third, integration with the existing document management workflow – the system must surface output within the tools writers and reviewers already use, rather than requiring them to adopt a separate platform. Beyond these technical prerequisites, involving medical writers in design and piloting from the outset is the single factor most consistently associated with successful real-world adoption.
How much time does an intelligent medical writing platform actually save when drafting a CSR?
The most precise validated data point available comes from Merck’s deployment with McKinsey, which reduced first-draft time from 180 hours to 80 hours – a 56% reduction in writer touch time – and compressed the calendar timeline from two to three weeks to three to four days. Broader McKinsey analysis across clinical development programmes reports roughly 40% CSR timeline acceleration as a representative benchmark. The savings concentrate in the data extraction and transcription phase rather than the prose drafting phase – which is why even partial automation of that upstream step produces large efficiency gains. These gains are strongest for high-volume, structurally consistent documents like patient narratives and data summary sections, and less pronounced for complex discussion and integrated summary sections that require substantial human scientific authorship regardless.
Is an AI medical writing tool secure enough to handle clinical trial patient data?
Security is fully addressable but requires a deliberate architectural decision at the outset. Routing clinical trial data containing patient identifying information through a general-purpose public API would violate applicable privacy regulations in most jurisdictions – this is not a viable approach for any responsible implementation. However, on-premise deployment or enterprise cloud environments configured with appropriate data processing agreements enable the system to operate entirely within the organisation’s controlled security perimeter. In this configuration, patient data never leaves the company’s own infrastructure. Audit trail and access control requirements for regulated pharmaceutical environments are equally manageable within GxP-compliant system architectures. Security in this context is a design and deployment decision – not an inherent limitation of the technology.
Build This AI Medical Writing Automation Solution With Softlabs Group
Softlabs Group builds custom AI medical writing automation solutions engineered to your organisation’s specific data environment, regulatory templates, and document management infrastructure. We do not configure generic tools to your use case. We build purpose-specific systems – retrieval-locked pipelines, on-premise or private cloud LLM deployments, GxP-validated audit trails, and Word and content management API integrations – designed from the ground up around how your writing team actually works. For pharmaceutical companies and CROs requiring a production-ready, inspection-safe automated regulatory writing software capability, our team brings both the AI engineering depth and the regulatory domain understanding to build it responsibly.
Whether you are building a first automated clinical documentation platform for pharma writers, or expanding an existing AI programme to cover additional document types, the starting point is a structured technical conversation. Explore our enterprise AI development capabilities for regulated industries, then reach out to begin that conversation with our regulatory AI specialists.